Apr
12
4:05 PM16:05
The workshop is built around four main topics:
Data Management and Standardized Data
Electronic Structure Methods and Derived Properties
Reliable Characterization and Modeling of Mesoscale Systems Bridging Length and Time Scales
Numerical Approaches Needed Towards Tailored MOF Systems for Real Applications

For each of the four topics, an overview of the current state of the art, perspectives and limitations will be given by a renowned scientist in the field.
prof. dr. Berend Smit (EPFL, Switzerland)
prof. dr. Aron Walsh (Imperial College Londen, UK)
prof. dr. Michele Parrinello (Istituto Italiano di Tecnologia, Italy)
prof. dr. Tom Woo (University of Ottowa, Canada)

The following invited speakers confirmed their presence:
prof. dr. Randall Snurr (Northwestern University, USA)
prof. dr. Stefan Kaskel (TU Dresden, Germany)
dr. Roberta Poloni (CNRS - SIMaP laboratory, France)
prof. dr. Silvia Bordiga (Università di Torino, Italy)
prof. dr. Volker Deringer (University of Oxford, UK)
dr. Simon Krause (Max-Planck-Institute for Solid State Research, Germany)
prof. dr. Seda Keskin (Koç University, Türkiye)
prof. dr. Norbert Stock (CAU, Germany)

The plenary discussions for the four topics will be moderated by
dr. Jack D. Evans (University of Adelaide, Australia)
prof. Egbert Zojer (Graz University of Technology, Austria)
Prof. dr. Özgür Yazaydin (University College London, UK)
dr. Peyman Z. Moghadam (University College London, UK)
SESSION 4: TOWARDS LONGER LENGTH SCALES
Laboratory of molecular simulation (LSMO), Institut des Sciences et Ingénierie Chimiques, Valais
Ecole Polytechnique Fédérale de Lausanne (EPFL), Rue de l’Industrie 17, CH-1951 Sion, Switzerland
Keywords: materials genomics, metal-organic frameworks, molecular qimulation, carbon capture, machine learning
The attractive feature of Metal Organic Frameworks (MOFs) isthat by changing the ligand and/or metal, theycan be chemically tuned to perform optimally for agiven application. This unique chemical tunability allows us to tailor-make materials that are optimal for a given application. The promise of finding just the right material seems remote however: because of practical limitations we can only ever synthesize, characterize, and test a tiny fraction of all possible materials. To take full advantage of this development, therefore, we need to develop alternative techniques, collectively referred to as Materials Genomics,[1] to rapidly screen large numbers of materials and obtain fundamental insights into the chemical nature of the ideal material for a given application. These computational materials genomics initiatives have been so successful that we have created a new problem: what to do with so much data? In this presentation we will discuss different computational strategies to dealwith a large amount of data. We illustrate on the use of these strategies by addressing the following questions: How the find the best material for a given application? How to find materials with similar pore shape?[2] How to design a material that optimally binds CO2? And, what can we learn from failed experiments?[3]
[1] P. G. Boyd, Y. Lee, and B. Smit, Computational development of the nanoporous materials genome, Nat. Rev. Mater., 2, 17037, 2017
[2] Y. Lee, S. D. Barthel, P. Dlotko, S. M. Moosavi, K. Hess, and B. Smit, Quantifying similarity of pore-geometry in nanoporous materials, Nat. Commun., 8, 15396, 2017
[3] S. M. Moosavi, A. Chidambaram, L. Talirz, M. Haranczyk, K. C. Stylianou, and B. Smit, Capturing chemical intuition in synthesis of metal-organic frameworks, Nat. Commun., 10, 539, 2019
SESSION 4: TOWARDS LONGER LENGTH SCALES
Department of Chemical Engineering, University College London, London, WC1E 7JE, United Kingdom
Mixed-matrix membranes (MMMs) are comprised of a continuous polymeric phase in which a carefully selected inorganic material is dispersed, changing the properties of the composite membrane, ideally to give higher permeability of the desirable gas species, and often simultaneously improving mechanical properties. In particular, MMMs which incorporate polymers and MOFs have been hailed for a wide range of membrane-based liquid and gas separation processes.[1-2] As such, fundamental understanding of the polymer/MOF interface/structure and of the fluid transport, whether in gas or liquid phase, through the composites is crucial to the rational selection of MOF/polymer pairs to achieve outstanding separation of mixtures. We recently developed a non-equilibrium molecular dynamics simulation method[3] in order to study thepermeation of pure fluids and separation of mixtures in membranes, namely "Concentration Gradient Driven Molecular Dynamics" (CGD-MD). This method works by employing bias forces to fix the concentration of fluids (pure or mixture) at the inlet and outlet of a membrane in order to create and maintain a concentration gradient across the membrane which drives the diffusion of molecules. This is aimed at providing a realistic representation of membrane separation experiments; for instance, high concentration/pressure at the feed side and vacuum at the permeate side. In this talk I’ll present our research on the application of the CGD-MD method to study gas transport and separation in MOF and composite MOF/Polymer membranes.
[1] B. Zornoza, C. Telleza, J. Coronasa, J. Gascon and F. Kapteijn, Metal organic framework based mixed matrix membranes: An increasingly important field of research with a large application potential, Micropor. Mesopor. Mater., 166, 67-68, 2013
[2] R. Semino, N. A. Ramsahye, A . Ghoufi and G. Maurin, Microscopic Model of the Metal−OrganicFramework/Polymer Interface: A First Step toward Understanding the Compatibility in Mixed Matrix Membranes, ACS Appl. Mater. Interfaces, 8, 809−819, 2016
[3] A. Ozcan, C. Perego, M. Salvalaglio, M. Parrinello, and O. Yazaydin, Concentration gradient driven molecular dynamics: a new method for simulations of membrane permeation and separation, Chem. Sci., 8, 3858-3865, 2017
SESSION 4: TOWARDS LONGER LENGTH SCALES
Institut Charles Gerhardt Montpellier/Université de Montpellier, Montpellier, France
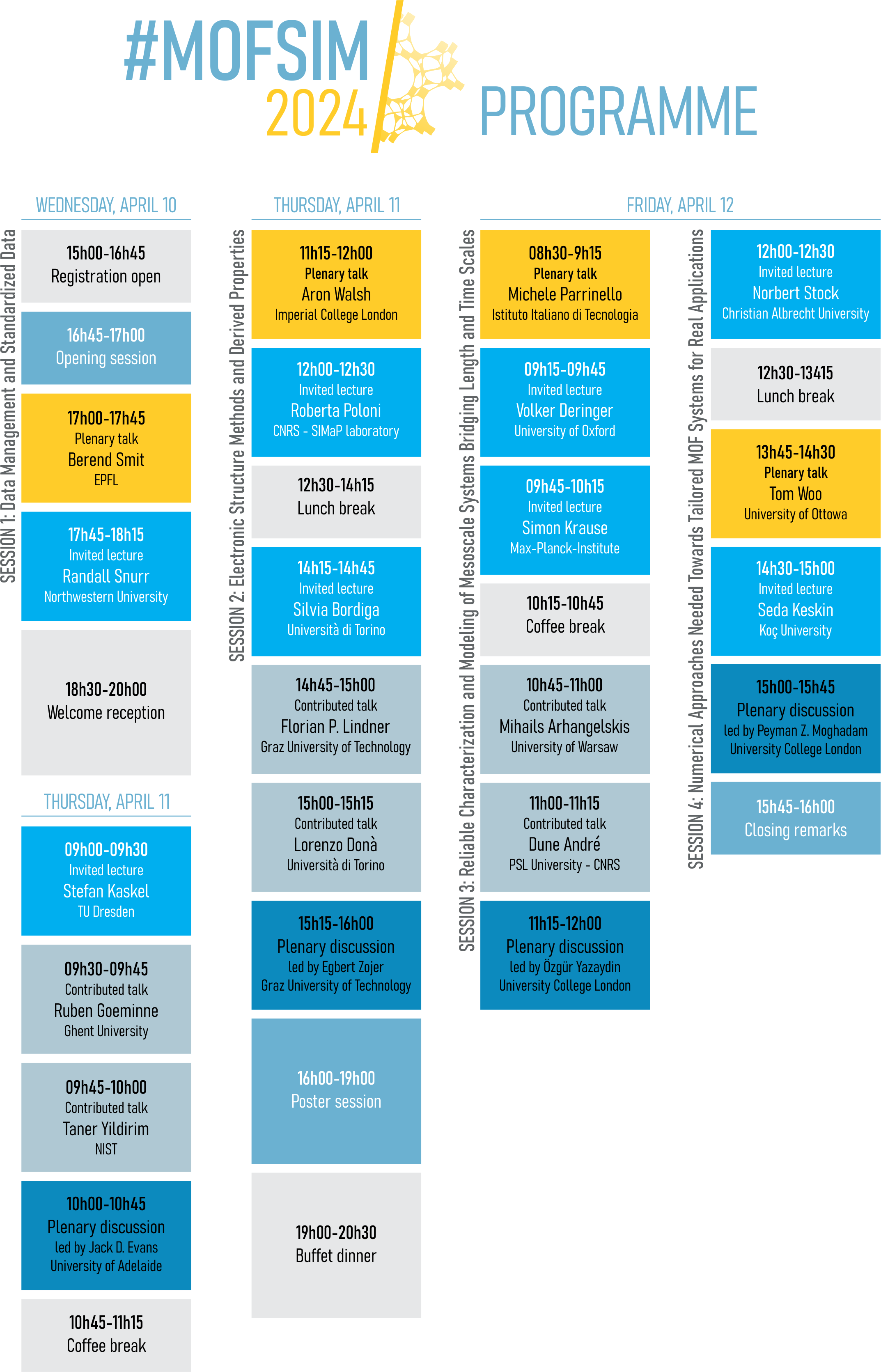
MOF/polymer Mixed Matrix Membranes (MMMs) have attracted great interest in the last few years as a promising alternative to the polymer membranes currently used for gas separation processes.[1] Although many experimental studies have focused on the elaboration and efficacy of such MMMs for specific gas separations, both the interfacial structure of these composites and the molecular mechanism of the separation phenomena are far from being understood. In this contribution, we first present a computational methodology to model the microscopic structural features of MOF/polymer interfaces.[2] Within our framework, the MOF surface and the polymer are modelled separately at the atomistic level and then combined through a series of molecular dynamics simulations to guarantee the local relaxation of the polymer structure in the presence of the chemical environments that constitute the MOF surface. We further investigate the full extent of the interfacial polymer by extending our analysis to longer time and length scales using a mesoscopic model,[3] based on a coarse grained MOF model recently proposed by Dürholt et al.[4](see Figure 1). By comparing our results with experimental findings and rationalizing them we have determined the key MOF and polymer characteristics that determine their compatibility within the binary composite.[5] Finally, we present some applications of our methodology to the study of complex phenomena such as the CO2 adsorption at the interface of the composites[6] and the polymer infiltration into the open pores at the MOF surface.[7]
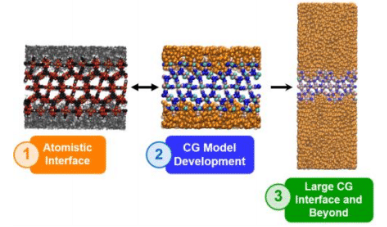
Figure 1: Summarized workflow of our multiscale study of a MOF/Polymer interface
[1] B. Seoane, J. Coronas, I. Gascón, M. E. Benavides, O. Karvan, J. Caro, F. Kapteijn, J. Gascón, Chem. Soc. Rev. 44, 2421, 2015
[2] R. Semino,N. Ramsahye,A. Ghoufi,G. Maurin, ACS Appl. Mater. Interfaces 8, 809, 2016
[3] R. Semino, J. P. Dürholt, R. Schmid, G. Maurin,J. Phys. Chem. C 121, 21491, 2017
[4] J. P. Dürholt, R. Galvelis, R. Schmid, Dalton Trans. 45, 4370, 2016
[5] R. Semino, J. C. Moreton, N. A. Ramsahye, S. M. Cohen, G. Maurin, Chem. Sci. 9, 315, 2018
[6] S. Hwang, R. Semino, B. Seoane, M. Zahan, C. Chmelik, R. Valiullin, M. Bertmer, J. Haase, F. Kapteijn, J. Gascon, G. Maurin, J. Kärger, Angew. Chem. Int. Ed. 57, 5156, 2018
[7] P. Duan, J. C. Moreton, S. R. Tavares, R.Semino, G. Maurin, S. M. Cohen, K. Schmidt-Rohr, submitted
SESSION 3: MECHANICAL, THERMAL, AND CHEMICAL STABILITY OF MOFS
Department of Chemistry, University of Torino, via Giuria 5, 10125, Torino, Italy
Keywords: elastic constants, quasi-harmonic approximation, shear instability, thermal expansion
Quantum-mechanical calculations based on the density functional theory (DFT) represent an effective mean to the determination and atomistic rationalization of many properties of materials. In recent years, much effort has gone in the development of effective and robust algorithms for the description of advanced properties of materials, and in their implementation into general-purpose public codes.
Here, I focus on the inclusion of pressure and temperature effects on structural, mechanical and thermodynamic properties of materials from DFT calculations. More specifically, I discuss recent progresses and open challenges of the application of such methodologies to the description of two classes of materials: molecular crystals and MOFs. These soft systems are held together by a variety of different chemical interactions (including weak ones), which need a balanced description, and are often characterized by alarge number of atoms per cell.
In this contribution, I review some of the methodologies that I have implemented into the public CRYSTAL program[1-2] to include pressure and thermal effects (beyond the harmonic approximation) on computedproperties of solids[3-4]. Examples will be given on the application of these methods to molecular crystals[5-6] and MOFs[7-9].

Figure 1. Left panel: Evolution with pressure of the mechanical anisotropy of ZIF-8. Right panel: structural andmechanical response of MOF-5 to pressure and temperature.
[1] R. Dovesi, A. Erba, et al., WIREs Comput. Mol. Sci., e1360, 2018
[2] A. Erba, J. Baima, et al., J. Chem. Theory Comput., 13, 5019, 2017
[3] A. Erba, J. Chem. Phys., 141, 124115, 2014
[4] A. Erba, A. Mahmoud, et al., J. Chem. Phys., 140, 124703, 2014
[5] A. Erba, J. Maul and B. Civalleri, Chem. Commun., 52, 1820, 2016
[6] M. Ruggiero, A. Zeitler and A. Erba, Chem. Commun., 53, 3781, 2017
[7] W. Zhang, J. Maul, et al., J. Phys. Chem. C, 122, 27442, 2018
[8] J. Maul, M.R. Ryder, et al., Phys. Rev. B., 99, 014102, 2019
[9] M.R. Ryder, J. Maul, et al., Adv. Theor. Simul., in preparation, 2019
SESSION 3: MECHANICAL, THERMAL, AND CHEMICAL STABILITY OF MOFS
Department of Chemical and Biological Engineering, The University of Sheffield, United Kingdom
Keywords: CSD MOF subset, adsorption simulations,operando molecular dynamics, MOF stability
The nature of MOFs has been under intense debate for some years, since, as defined by the IUPAC in the MOF 2012 conference, “a MOF is what looks like a MOF”. This debate has been exacerbated with the emergence of new MOFs with complex structural chemistries – something that continuously increases with advances in synthesis methods. We aimed to answer these challenges by investigating the nature of the question “what is a MOF?” and creating structure search methods within the Cambridge Structural Database (CSD) through a number of “look-for-MOF” criteria to identify and create a curated database of ca. 88,000 MOF materials to date.[1] The existence of this many structures clearly creates a lot of opportunities, but it also brings the challenge of how does one identify a promising MOF for a particular application among all these structures? Here, we highlight two research projects where computational high-throughput screening has been employed to design and discover MOFs. The first project presents a rare case of computer-aided MOF discovery, in which we complete the full cycle from rapid high-throughput computational screening of MOF materials for oxygen storage, to identification, synthesis and measurement of oxygen adsorption in the top-ranked structure.[2] In the second project, we employ a multi-level computational approach to delineate key structural features in MOFs that influence their mechanical properties. The results guide MOF researchers to assess and design structures with improved mechanical stability.[3]

Fig.1. Structure-property relationships for oxygen storage[2] (left) and mechanical properties[3] (right)
[1] P. Z. Moghadam, A. Li, S. B. Wiggin, A. Tao, A. G. P. Maloney, P. A. Wood, S. C. Ward, and D. Fairen-Jimenez. Chem. Mater., 29, 2618–2625, 2017
[2] P. Z. Moghadam, T. Islamoglu, S. Goswami, J. Exley, M. Fantham, C. F. Kaminski, R. Q. Snurr, O. K. Farha, and D. Fairen-Jimenez, Nat. Commun., 9, 1-8, 2018
[3] P. Z. Moghadam, S. M. J. Rogge, A. Li, C. Chow, J. Wieme, N. Moharrami, M. Aragones-Anglada, G. Conduit, D. A. Gomez-Gualdron, V. Van Speybroeck, D. Fairen-Jimenez, Matter, accepted, 2019
SESSION 3: MECHANICAL, THERMAL, AND CHEMICAL STABILITY OF MOFS
Multifunctional Materials & Composites (MMC) Lab, Department of Engineering Science, University of Oxford, United Kingdom
Keywords: elastic anisotropy, nanoindentation, vibrational spectroscopy, density functional theory, terahertz modes
Understanding the mechanical properties of MOF crystals and the structure-property relations of new compounds is central to every practical applications. This talk will discuss exemplars derived from our studies of this fast-expanding topic area. I shall begin by discussing the quasistatic mechanical properties of single crystals of MOF. Example will be drawn from the accurate determination of the elastic constants (Cij’s) of a micron-size ZIF-8 crystal[1], measured using a combination of Brillouin spectroscopy and nanoindentation methods, and further validated using ab initiodensity functional theory (DFT) calculations via CRYSTAL14. Recently, we made progress in the nanoindentation measurements of sub-micron crystals of ZIF-8 employing an atomic force microscope (AFM) approach[2], which provides the unique opportunity to measurethe mechanical behaviour of a MOF crystal (500 nm – 1 µm) which is around 500 times smaller than those employed in [1].In the second part of the talk, I will address the dynamic mechanical response of MOF structures, focussing on the study of co-operative THz vibrations using a combination of synchrotron far-IR spectroscopy, inelastic neutron scattering and DFT techniques [3]. It is proposed that connections exist between the anisotropic elastic properties of the open porous framework and their collective vibrational motions (rotor dynamics[4] and linker buckling) to attain anomalous elastic response such as negative Poisson’s ratio (auxeticity)[5]. Finally, I shall discuss the liquid intrusion phenomenon into ZIFs subject to a moderate mechanical pressure of tens of MPa, where reversible liquid intrusion-extrusion response has been detected accompanied by rate-dependent deformation effects—indicative of a creep/stress relaxation type mechanism[6,7].
[1] Tan, J.C., Civalleri, B., Lin, C.C., Valenzano, L., Galvelis, R., Chen, P.F., Bennett, T.D., Mellot-Draznieks, C., Zicovich-Wilson, C.M. and Cheetham, A.K., Exceptionally Low Shear Modulus in a Prototypical Imidazole-Based Metal-Organic Framework, Phys. Rev. Lett., 108, 095502, 2012
[2] Zeng, Z. and Tan, J.C., AFM Nanoindentation To Quantify Mechanical Properties of Nano-and Micron-Sized Crystals of a Metal-Organic Framework Material, ACS Appl. Mater. Interfaces, 9, 39839, 2017
[3] Ryder, M.R., Civalleri, B., Bennett, T.D., Henke, S., Rudić, S., Cinque, G., Fernandez-Alonso, F. and Tan, J.C., Identifying the Role of Terahertz Vibrations in Metal-Organic Frameworks: From Gate-Opening Phenomenon to Shear-Driven Structural Destabilization, Phys. Rev. Lett., 113, 215502, 2014
[4] Ryder, M.R., Van de Voorde, B., Civalleri, B., Bennett, T.D., Mukhopadhyay, S., Cinque, G., Fernandez-Alonso, F., De Vos, D., Rudic, S. and Tan, J.C., Detecting Molecular Rotational Dynamics Complementing the Low-Frequency Terahertz Vibrations in a Zirconium-Based Metal-Organic Framework, Phys. Rev. Lett., 118, 255502, 2017
[5] Tan, J.C., Civalleri, B., Erba, A. and Albanese, E., Quantum mechanical predictions to elucidate the anisotropic elastic properties of zeolitic imidazolate frameworks: ZIF-4 vs. ZIF-zni, CrystEngComm, 17, 375, 2015.
[6] Sun, Y., Li, Y. and Tan, J.C., Liquid Intrusion into Zeolitic Imidazolate Framework-7 Nanocrystals: Exposing the Roles of Phase Transition and Gate Opening to Enable Energy Absorption Applications, ACS Appl. Mater. Interfaces, 10, 41831, 2018
[7] Sun, Y., Li, Y. and Tan, J.C., Framework flexibility of ZIF-8 under liquid intrusion: discovering time-dependent mechanical response and structural relaxation, Phys. Chem. Chem. Phys., 20, 10108, 2018
SESSION 3: MECHANICAL, THERMAL, AND CHEMICAL STABILITY OF MOFS
Ruhr-University Bochum, Computational Materials Chemistry group, Bochum, Germany
Keywords: pillared layer MOFs, force field simulations, phase transition
By adding flexible chains to the linkers, pillared layer MOFs can be converted to breathing and gating systems. For the theoretical investigation of stimuli induced phase transitions it is advantageous to focus on the temperature induced opening of such systems. Here no grand canonical system with a changing number of guest molecules needs to be simulated, as it would be the case for adsorption induced transformations. Previously we have used our first-principles parameterized force field MOF-FF[1] to investigate this process within periodic boundary conditions[2]. However, in reality an interface between phases can form, which is only to be realized in a simulation system ofvery large size, or, ideally, without the artificial constraint of periodic boundary conditions for a nanoparticle.In the presentation a revised MOF-FF for these systems will be presented, based on a force matching for the flexible side chains. By extending the numerically efficient lammps code with the potential terms necessary for MOF-FF substantially larger systems can now be investigated. Our first results on the thermal opening of nanoparticles of different size will be discussed.

Figure 1: Opening of a 6x6x6 BME-fu-MOF nanoparticle (Cu(BME-bdc)2(dabco), BME-bdc: 2,5-bismethoxyethoxy-benzenedicarboxylate) by heating from 300K to 500K.
[1] S. Bureekaew, S. Amirjalayer, M. Tafipolsky, C. Spickermann, T. K. Roy, R. Schmid, Phys. Stat. Sol. B, 250, 1128-1141, 2013
[2] M. Alaghemandi, R. Schmid, J. Phys. Chem. C, 120, 6835-6841, 2016
SESSION 3: MECHANICAL, THERMAL, AND CHEMICAL STABILITY OF MOFS
Chimie ParisTech, PSL University, CNRS, France
Keywords: defects, disorder, porous materials, multi-scale modelling
Recent years have seen a large increase of the research effort focused on framework materials, including the nowadays-ubiquitous metal–organic frameworks, but also dense coordination polymers, covalent organic frameworks, and molecular frameworks. A large number of these frameworks flexible, or stimuli-responsive, and there is growing evidence that large-scale flexibility, the presence of defects and long-range disorder are not the exception in metal-organic frameworks, but the rule.Our group has put together a “toolbox” of theoretical approaches to shed light into these materials’ properties, and in particular to understand their behavior under mechanical constraints and temperature changes, the interplay between the phenomena of adsorption, deformation and reactivity of these materials, and their optical properties. By means of molecular simulation at varying scale, we can now probe, rationalize and predict the behavior of stimuli-responsive materials, producing a coherent description of Soft Porous Crystals from the unit cell scale all the way to the behavior of the whole crystal.This is particularly important in understanding the links between flexibility, defects and disorder, all of which arise from the large dimensionality of these complex supramolecular assemblies.

[1] M. Baise, P. M. Maffettone, F. Trousselet, N. P. Funnell, F.-X. Coudert and A. L. Goodwin, Negative Hydration Expansion in ZrW2O8: Microscopic Mechanism, Spaghetti Dynamics, and Negative Thermal Expansion, Phys. Rev. Lett., 120, 265501, 2018
[2] R. Gaillac, P. Pullumbi, K. A. Beyer, K. W. Chapman, D. A. Keen, T. D. Bennett and F.-X. Coudert, Liquid metal-organic frameworks, Nat. Mater., 16, 1149–1154, 2017
[3] T. D. Bennett, A. K. Cheetham, A. H. Fuchs and F.-X. Coudert, Interplay between defects, disorder and flexibility in metal-organic frameworks, Nat. Chem., 9, 11–16, 2017
[4] A. B. Cairns, M. J. Cliffe, J. A. M. Paddison, D. Daisenberger, M. G. Tucker, F.-X.Coudert and A. L. Goodwin, Encoding Complexity within Supramolecular Analogues of Frustrated Magnets, Nat. Chem., 8, 442–447, 2016
SESSION 2: ELECTRONIC PROPERTIES AND DERIVED FUNCTIONS OF MOFS
Delft University of Technology, Department of Chemical Engineering, Delft, The Netherlands
Keywords: disorder, dynamics, structure
It may be tempting to think of the structure of many crystalline materials as perfectly crystalline and static. This is far from the truth. There is growing evidence that long-range disorder and defects are fairly common in metal-organic frameworks, and affect the materials’ functionalities.[1] In addition, these frameworks are highly dynamic. Rotational dynamics of linkers for example are very prevalent, and should be taken into account in modelling of many of the materials’ properties.[2] Determining the disorder and dynamics, however, is all but trivial. A combined experimental and theoretical approach can greatly aid here, as I’ll show with a few examples. The talk will focus on disorder in linker organisation, rotational dynamics of linkers and the piezoelectric response of examples from the ZIF and MIL-53 family.
[1] T.D. Bennett, A.K. Cheetham, A.H. Fuchs, F.-X. Coudert, Nature Chem. 9, 11, 2017
[2] A.G. Nelson, F.-X. Coudert, M.A. van der Veen, Nanomaterials 9, 330, 2019
SESSION 2: ELECTRONIC PROPERTIES AND DERIVED FUNCTIONS OF MOFS
Graz University of Technology, Institute of Physical and Theoretical Chemistry, Austria
The University of Adelaide, Department of Chemistry, Australia
Keywords: self-assembly, mineralization, biomolecule, MOF composite, bio-MOF
Among the different classes of Metal-organic Framework (MOF) composites prepared during recent years using ceramic, metallic and polymeric nanoparticles,[1-4] a newe merging type of MOF composite has been recently obtained encapsulating bio-macromolecules within MOFs.[5-7] In physiological solutions,co-precipitation and biomimetic mineralization methods have been used to self-assemble MOFs around bio-active compounds (e.g., enzymes). The biomimetic mineralization of MOF bio-composites enables the fast encapsulation of guests larger than micropores of MOFs.[8] This new class of bio-composites have shown unprecedented properties for biotechnological applications.[9] In this presentation, we will discuss about different biomacromolecules (e.g., proteins, carbohydrates) and complex biological systems (yeast cells) as crystallization agents for MOFs.[10–13] The functional properties of these composites will be disclosed providing examples of other methods used for the encapsulation of proteins within MOFs, including the preparation of hollow MOF capsules.[14,15] Exciting potential applications of these new MOF bio-composites and current challenges will be presented.[8,16]
[1] P. Falcaro, R. Ricco, A. Yazdi, I. Imaz, S. Furukawa, D. Maspoch, R. Ameloot, J. D. Evans and C. J. Doonan, Coord. Chem. Rev. 307, 237–254, 2016
[2] Q.-L. Zhu and Q. Xu, Chem. Soc. Rev. 43, 5468–5512, 2014
[3] C. M. Doherty, D. Buso, A. J. Hill, S. Furukawa, S. Kitagawa and P. Falcaro, Acc. Chem. Res., 47, 396–405, 2014
[4] G. Li, H. Kobayashi, J. M. Taylor, R. Ikeda, Y. Kubota, K. Kato, M. Takata, T. Yamamoto, S. Toh, S. Matsumura and H. Kitagawa, Nat. Mater., 13, 802–806, 2014
[5] F. Lyu, Y. Zhang, R. N. Zare, J. Ge and Z. Liu, Nano Lett., 14, 5761–5765, 2014
[6] K. Liang, R. Ricco, C. M. Doherty, M. J. Styles, S. Bell, N. Kirby, S. Mudie, D. Haylock, A. J. Hill, C. J. Doonan and P. Falcaro, Nat. Commun., 6, 7240, 2015
[7] F.-K. Shieh, S.-C. Wang, C.-I. Yen, C.-C. Wu, S. Dutta, L.-Y. Chou, J. V. Morabito, P. Hu, M.-H. Hsu, K. C.-W. Wu and C.-K. Tsung, J. Am. Chem. Soc., 137, 4276–4279, 2015
[8] R. Riccò, W. Liang, S. Li, J. J. Gassensmith, F. Caruso, C. Doonan and P. Falcaro, ACS Nano, 12, 13–23, 2018
[9] C. Doonan, R. Riccò, K. Liang, D. Bradshaw and P. Falcaro, Acc. Chem. Res., 50, 1423–1432, 2017
[10] N. K. Maddigan, A. Tarzia, D. M. Huang, C. J. Sumby, S. G. Bell, P. Falcaro and C. J. Doonan, Chem. Sci., 9, 4217–4223, 2018
[11] W. Liang, H. Xu, F. Carraro, N. K. Maddigan, Q. Li, S. G. Bell, D. M. Huang, A. Tarzia, M. B. Solomon, H. Amenitsch, L. Vaccari, C. J. Sumby, P. Falcaro and C. J. Doonan, J. Am. Chem. Soc., 141, 2348–2355, 2019
[12] E. Astria, M. Thonhofer, R. Ricco, W. Liang, A. Chemelli, A. Tarzia, K. Alt, C. E. Hagemeyer, J. Rattenberger, H. Schroettner, T. Wrodnigg, H. Amenitsch, D. M. Huang, C. J. Doonan and P. Falcaro, Mater. Horiz., 2019. DOI: 10.1039/C8MH01611A
[13] K. Liang, J. J. Richardson, J. Cui, F. Caruso, C. J. Doonan and P. Falcaro, Adv. Mater., 28, 7910–7914, 2016
[14] J. Huo, J. Aguilera-Sigalat, S. El-Hankari and D. Bradshaw, Chem. Sci., 6, 1938–1943, 2015
[15] G.-Y. Jeong, R. Ricco, K. Liang, J. Ludwig, J.-O. Kim, P. Falcaro and D.-P. Kim, Chem. Mater., 27, 7903–7909, 2015
[16] K. Liang, C. Carbonell, M. J. Styles, R. Ricco, J. Cui, J. J. Richardson, D. Maspoch, F. Caruso and P. Falcaro, Adv. Mater., 27, 7293–7298, 2015
SESSION 2: ELECTRONIC PROPERTIES AND DERIVED FUNCTIONS OF MOFS
School of Science, Chair of Theoretical Chemistry, TU Dresden, 01062 Dresden, Germany
Helmholtz Center Dresden-Rossendorf, Institute of Resource Ecology, Leipzig Research Branch, Permoserstr. 15, 04318 Leipzig, Germany
Keywords: hydrogen, isotopes, nuclear quantum effects, host-guest interactions, isotope separation
A lot of research has been devoted to Metal-Organic Frameworks (MOFs) as potential candidates for room temperature hydrogen storage. Optimistic predictions, in particular fuelledbyclassical computersimulations, have pushed the fieldforward in the 2000’s. However, due tolarge discrepancy to experiment, which failed confirming those predictions, the research field collapsed. Recently, we observe a renaissance ofhydrogen storage at liquid nitrogen temperature (77 K). In this talk, I will first highlight why most classical computer simulations on hydrogen in MOFs are flawed. I will introducea quantum-mechanical method to treat correctly the surface-adsorbed hydrogen quantum liquid. Then, I will focus on the treatment of nuclear quantum effects at strongly adsorbing sites in MOFs.In the last part of the presentation I will demonstrate that the nuclear quantum effects of hydrogen can be exploited for hydrogen isotope separation, in particular for the capture of radioactive tritium.
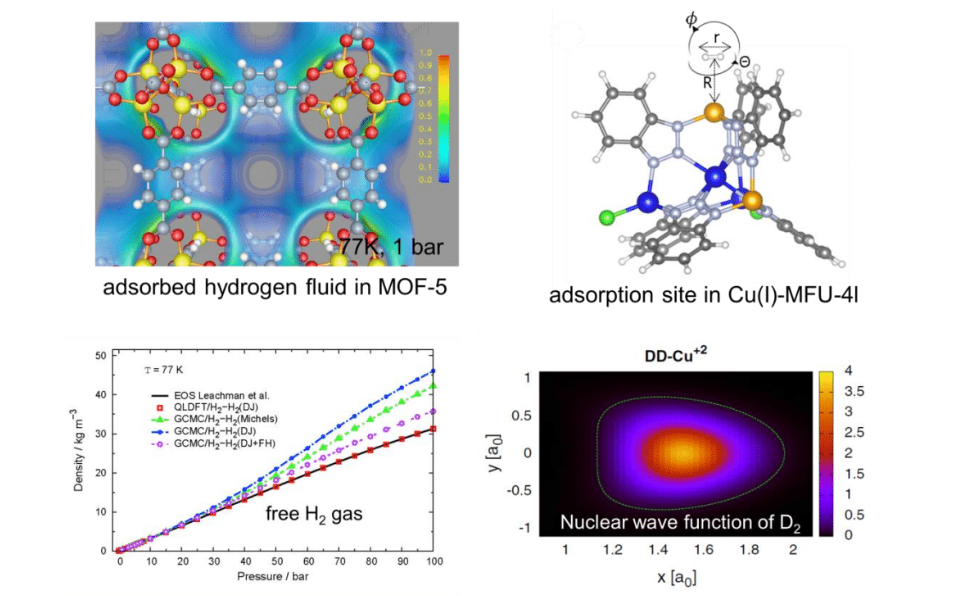
Figure 1: Some examples showing the importance of nuclear quantum effects of adsorbed dihydrogen.
SESSION 2: ELECTRONIC PROPERTIES AND DERIVED FUNCTIONS OF MOFS
Department of Chemistry, University College London, London WC1H 0AJ , United Kingdom
Keywords: defect, DFT, catalysis, vacancy
In this talk I will endeavour to summarise our efforts to provide a basic insight into defect formation energetics and characterisation of defects in MOFs, in the crystal interior and exterior. I will focus chiefly on UiO-66, mainly because defects are key to the catalytic behaviour of this material. As has been well documented, UiO-66 can tolerate exceptionally high concentrations of missing linker defects, where modulators such as formic acid replace the benzene dicarboxylate (BDC) groups. The nature of the acid sites associated with the defects has been disputed [1,2,3] but theory provides a consistent prediction of the nature of the defect centres, especially in the case when hydroxide acts as the capping/charge compensating species. Whilst the local structure of point defects in UiO-66 are well understood, the distribution of point defects and “missing cluster” defects as reported by Cliffe et al.[4] is less clear. Cliffe et al. postulated and produced evidence from diffuse scattering experiments which suggests that ordered missing cluster defects, corresponding to a nanodomain of reotopology, within the perfect UiO-66 fcutopology. I will show that we have been able to shed more light on the integrity of UiO-66 through a combination of theory, HRTEM and catalytic test reactions. Our work reveals that defective UiO-66 consists of fcu, reo and two newly identified nanodomains of bcu and scu. Intriguingly, the evolution of these nanodomains has been followed in two different modes of crystal growth: moderate concentration of modulator (50:1, formate: BDC)and high concentration of modulator (150:1, formate: BDC). In the former case, Ostwald ripening is observed and the crystals mature, containing a distribution of fcu, reo, bcu and scu to tend towards perfect fcu. The latter results in further ligand exchange resulting in increasing concentration of scu. Perhaps most interestingly, through performing catalytic test reactions at different points in the genesis of UiO-66, we make a clear distinction between their activity of UiO-66 that contains missing linkers only and that which also contains missing clusters. We find evidence that missing clusters are demonstrably more reactive than missing linkers, which points to the use of high modulator concentrations to engineer in potent active sites.
[1] Caratelli, C et al., J. Catal. 352, 401-414, 2017
[2] Ling, S. and Slater, B. Chem. Sci. 7, 4706-4712, 2016
[3] Trickett, C.A. et al., Angew. Chem., Int. Ed. 54, 11162-11167, 2015
[4] Cliffe, M.J. et al., Nat. Commun. 5, 4176, 2014
SESSION 2: ELECTRONIC PROPERTIES AND DERIVED FUNCTIONS OF MOFS
Department of Chemistry and Biochemistry, Materials Science and Engineering, San Diego Supercomputer Center, University of California, San Diego, La Jolla, CA 92093, U.S.A.
Keywords: water adsorption, confined water, proton conduction, framework flexibility, infrared spectroscopy
Metal-organicf rameworks have recently attracted much interest as promising materials for water generation and purification, proton conduction, and molecular sensing. Contrary to other porous materials like zeolites, MOFs are highly tunable, which implies that it is possible to rationally design the framework properties, including both pore size and shape, for specific applications. In this talk, I will present our results from advanced molecular dynamics simulations that are used to develop a microscopic picture of the mechanisms associated with water adsorption and water-mediated proton conduction in different MOFs. Particular focus will be on the analysis of both guest and framework properties as a function of temperature, guest loading, and pore size/shape, which are characterized in terms of specific spectroscopic signatures.
SESSION 1: CATALYSIS AND ADSORPTION IN MOFS
Delft University of Technology, P&E department, Leeghwaterstraat 39, 2628CB Delft, The Netherlands
Keywords: M-MOF-74, adsorption, polarizable force field
The separation of light olefins from paraffins via cryogenic distillation is a very energy intensive process. Solid adsorbents and especially metal–organic frameworks with open metal sites have the potential to significantly lower the required energy. Specifically, M-MOF-74 has drawn considerable attention for application in olefin/paraffin separation. To investigate how the separation proceeds on a molecular level and to design better materials, molecular simulation can be a useful tool. Unfortunately, it is still a challenge to model the adsorption behavior of many adsorbates in metal–organic frameworks with open metal sites. Previously, the inclusion of explicit polarization has been suggested to improve the quality of classical force fields for such systems. Here, the potential of polarizable force fields for the description of olefins and paraffins in metal–organic frameworks with open metal sites is investigated. In particular, heats of adsorption, binding geometries, and adsorption isotherms are calculated for C2H4, C2H6, C3H6, and C3H8 in M-MOF-74 (with M = Co, Mn, Fe, and Ni). In this study, no force field parameters are adjusted to improve the model. The results show that including explicit polarization significantly improves the description of the adsorption in comparison to non-polarizable generic force fields which do not consider explicit polarization. The study also reveals that simulation predictions are sensitive to the assigned repulsive potential and framework charges.
SESSION 1: CATALYSIS AND ADSORPTION IN MOFS
Center for Molecular Modeling, Ghent University, Technologiepark 46, Zwijnaarde 9052, Belgium
Keywords: soft porous crystals, negative gas adsorption, free energy, mechanical pressure, adsorption isotherm
Metal-organic frameworks (MOFs) are among the most intriguing materials of current science. Even though MOFs are crystalline, some have shown flexible behavior in the sense that they are capable of transforming between various phases accompanied by substantial changes in the unit cell volume. Kitagawa coined the term “soft porous crystals” (SPC) for materials that show a bistable or multistable behavior with long–range structural order and permanent porosity [1]. These SPCs are known for their intriguing properties and various counterintuitive phenomena such as negative linear compression, negative thermal expansion and negative gas adsorption (NGA). The latter was illustrated by the adsorption of methane in DUT-49 for which experimentally a drop in the amount of adsorbed methane was observed under increasing vapor pressure [2]. Recently, we proposed a generalized thermodynamic approach to classify SPCs into various types of flexibility upon exposure to mechanical pressure based on the Helmholtz free energy of the empty materials [3]. In the current work, we construct the Helmholtz free energy by means of a semi-analytical thermodynamic model[4] and build further upon this classification to investigate whether there is a correlation between pressure-induced breathing of the empty framework and adsorption-induced breathing at fixed mechanical pressure. As such we aim to determine the conditions required for NGA and investigate the correlation with pressure-induced breathing.
Figure 1 illustrates the adsorption isotherm for methane in a hypothetical non-breathing MOF (i.e., non-breathing in absence of mechanical pressure) for increasing values of an applied mechanical pressure, showing that increasing the mechanical pressure can induce NGA. Therefore, even though NGA was experimentally observed for methane adsorption in DUT-49, we herein discovered that it could be a more generally applicable phenomenon under influence of guest adsorption and external mechanical pressure, evenindicating that NGA might become tuneable. Although it is very difficult to perform the required experiments to test such statements with the currently available experimental setups, the present results still allows us to gain crucial insight into the NGA phenomenon. Furthermore, such meticulous control of multiple triggers for NGA can open the way to new applications such as tuneable gas detection and pressure amplification.
Figure 1: Illustration of the impact of applying mechanical pressure onthe adsorption isotherm of an initially non-breathing MOF. The blue and red dots represent the number of molecules adsorbed in the open pore and contracted pore state respectively.
[1] Horike, S., Shimomura, S. and Kitagawa, S.,Nat. Chem. 1, 695, 2009
[2] Krause, S. et al., Nature 532, 348, 2016
[3] Vanduyfhuys, L. et al., Nat. Commun. 9, 204, 2018
[4] Vanduyfhuys, L. et al., Mol. Simulat. 41, 1311–1328, 2015
SESSION 1: CATALYSIS AND ADSORPTION IN MOFS
Instituto de Tecnología Química, UPV-CSIC, Valencia (Spain)
Keywords: molecular dynamics, gate adsorption, water, metal-organic framework
Metal-organic frameworks (MOFs) have been many times pointed out as adsorbants displaying the so called gate-driven adsorption mechanism. Such mechanism implies, most of the times, two localised parts in the MOF with differentiated mechanical resistances, the harder –well extended– provides the robust framework required for structural integrity, and the softer –more localised– presents the mobility (gate) that is triggered by the adsorbate diffusion.
How selective are these gates is a matter of considerable importance in order to assess the MOF performance in separation processes. But, separation does not require gates, as exemplified by zeolites, active carbons, organic polymeric membranes, as well as the vast majority of MOFs whose separation ability does not rely on the presence of gates. The question arising is, what is –if any– the unique aspect of separation based on gates?
The structural peculiarities of MFU-4 (metal-framework Ulm number 4) feature a very robust framework containing an interconnected system of alternating large (11.9 Å) and small (2.5 Å) spherical cavities making a octahedrally (6:6) connected micropore. Small pores are very flexible and originate a unique 6-entrance gate of cubic shape made of 8 chlorine ligands (edge = 4.4 Å) interacting through van der Waals interactions. The flexibility of the small pore arises from the weak Cl---Cl interactions, allowing any vertex (Cl) to move, producing a large distortion leading to gate opening.
In previous studies[1,2],different cases of chemical interactions,with CO2, N2 and Xe adsorbates, were studied. The present study will illustrate how water leads to a new way of interaction with specific consequences on diffusion and uptake.
[1] Sastre, van den Bergh, Kapteijn, Denysenko, Volkmer, Unveiling the mechanism of selective gate-drivendiffusion of CO2 over N2 in MFU-4 metal–organicframework, Dalton Trans., 43, 9612, 2014
[2] Bunzen, Kolbe, Kalytta-Mewes, Sastre, Brunner, Volkmer, Achieving Large Volumetric Gas Storage Capacity in Metal−Organic Frameworks by Kinetic Trapping: Xenon Loading in MFU-4, J. Am. Chem. Soc. 140, 10191, 2018
SESSION 1: CATALYSIS AND ADSORPTION IN MOFS
c-MACS - Centre for Membranes, Adsorption, Catalysis and Spectroscopy, KULeuven
Keywords: Ti-MOF, Ce-MOF, MOF-808, bio-oil separation, catalytic C-H activation
In a first part of the lecture, we will explore the structure and reactivity of novel MOFs based on tetravalent cations, such as Ti4+, Ce4+ and Zr4+. We will elucidate the structure of a Ti-MOF material consisting of Ti4+-O2--Ti4+ chains, linked by 4,4’-biphenyldicarboxylate (bpdc) linkers. The eventual structural elucidation was only possible using a combination of advanced electron diffraction and solid state NMR. The redox photochemistry of the material will be discussed, and due to its high number of open coordination sites on Ti4+, the material is also an excellent catalyst for the oxidation of aromatic thiophenes. Next we will address new insights in the chemistry of Ce MOFs. While the Ce-UiO-66 material was originally reported to catalyze the TEMPO-mediated alcohol oxidation, we will now show that similar materials can also mediate redox processes of nitrogen oxides.
The chemistry of the Zr centres in UiO-66 and MOF-808 continues to be a rich source of inspiration in catalysis and adsorption. For instance, MOF-808 is one of the most active catalysts for the oxidation of aromatic thiophenes, and provided its polarity properties are well adjusted, it can also be used to catalyze epoxidation reactions.[1] MOF-808 is a particularly useful material in complex separations of biobased compounds, like encountered in bio-oil: due to its large number of open coordination sites onthe Zr clusters, MOF-808 selectively adsorbs guaiacols (2-methoxyphenols) and dihydroxybenzenes from a large variety of other compounds, including furanics, alcohols, sugars, acids etc.[2] Besides these applications that exploit the intrinsic reactivity of MOF framework atoms, MOFs can also be considered as one of the most powerful platforms to design immobilized homogeneous catalysts. We will illustrate this with the currently highly topical class of C-H activation reactions. The overall aim here is to find shortcuts to the currently established reactions like the Suzuki, Heck or Buchwald amination reactions. While the latter use pre-oxidized arene reactants (e.g., bromoarenes, iodoarenes) in combination with Pd catalysts, there is an interest to catalytically activate C-H bonds, rather than C-Br or C-I bonds. Such reactions are possible with Pd as well, but the reaction cycle then starts with Pd2+, which might act as an electrophile, but can also activate an arene C-H via a concerted-metallation deprotonation.
A first catalyst that will be discussed is a material allowing the dehydrogenative coupling of 2 arenes to form biaryl compounds. For instance, coupling of ortho-xylene results in bis-xylyl, which is a direct precursor in the production of the high performance Upilex polyamide material. We will show that Pd2+ can be directly grafted on uncovered Zr6 clusters of MOF-808, resulting in a three-fold increase of the lifetime of the Pd catalyst.[3] Alternatively, we will demonstrate that the building brick of MOF-808 can as well be used to dock carboxylic organic ligands –as an example we will present a tetrahydrothiophene-2,5-dicarboxylic acid, which as a ligand can support the catalytic activity of Pd2+ in a variety of C-H activating reactions.
[1] Fu, Bueken, De Vos et al., Small Methods 2, 1800203, 2018
[2] Jia, De Vos et al., ChemSusChem 12, 1256, 2019
[3] Van Velthoven, De Vos et al., Chem. Sci. 10, 3616, 2019
SESSION 1: CATALYSIS AND ADSORPTION IN MOFS
Department of Chemistry, Chemical Theory Center, and Supercomputing Institute, University of Minnesota, Minneapolis, Minnesota 55455-0431, United States
Keywords: catalysis, density functional, multireference methods, oxidation, oligomerization
Metal-organic frameworks (MOFs) are versatile platforms with tunable properties ranging from high selectivity in gas separations, to catalytic activity for complex reactions, to unique magnetic properties. In the Inorganometallic Catalyst Design Center, we combine theory and experiment to understand the activity of transitionmetal catalysts supported on MOF nodes for reactions related to natural gas conversion, e.g., catalytic oligomerization of abundant C1, C2, and C3 hydrocarbons to longer congeners, or their selective oxidation to alcohols or other fuel molecules. Modeling these species can pose enormous challenges from a theoretical and computational perspective.[1] I will describe our latest results for C–H bond activation by bimetallic oxide clusters deposited on Zr-based MOF nodes[2] and light alkane hydroxylation over Fe-based MOFs.[3]
[1] V. Bernales, M. A. Ortuño, D. G. Truhlar, C. J. Cramer, and L. Gagliardi, Computational Design of Functionalized Metal–Organic Framework Nodes for Catalysis, ACS Cent. Sci., 4, 5-19, 2018
[2] M. C. Simons, M. A. Ortuño, V. Bernales, C. J. Cramer, A. Bhan, and L. Gagliardi, C–H Bond Activation on Bimetallic Two-Atom Co-M Oxide Clusters Deposited on Zr-Based MOF Nodes: Effects of Doping at the Molecular Level, ACS Cat., 8, 2864–2869, 2018
[3] J. G. Vitillo, A. Bhan, C. J. Cramer, C. C. Lu and L. Gagliardi Quantum Chemical Characterization of Structural Single Fe(II) Sites in MIL-Type Metal Organic Frameworks for Oxidation of Methane to Methanol and Ethane to Ethanol, ACS Catal., 9, 2870–2879, 2019